후천성 혈우병 A
Acquired Hemophilia A
Article information
Trans Abstract
Acquired hemophilia A is rare but fatal bleeding disorder caused by an autoantibody against factor VIII. Acquired hemophilia A is more prevalent in the elderly patients and associated with other underlying diseases such as autoimmune disorders, malignancies, pregnancy, infection and several drugs. The bleeding pattern of acquired hemophilia A is quite different from that of congenital hemophilia A. Thus, acquired hemophilia A should be suspected in the presence of bleeding with sudden onset, often severe, which occurs spontaneously or after minor trauma, following invasive procedures or surgical interventions in patients without a personal or family history of bleeding. The main goal of the treatment for acquired hemophilia A should be the control of bleeding with the use of bypassing agents and the eradication of autoantibodies with the use of immunosuppressive agents. Because the remained level of factor VIII is not correlated with the risk of fatal bleeding, appropriate hemostatic management should be applied in patients with clinically significant bleeding regardless of the level of factor 8 or the inhibitor level of factor VIII. Because current recommendations for acquired hemophilia A are developed mainly based on the retrospectively available data, larger multicenter prospective trials should be conducted for developing appropriate treatments.
서론
후천성혈우병 A는 8번 응고인자에 대한 자가면역항체가 발생하여 혈중 8번 응고인자가 억제되고 고갈되어 출혈과 관련된 합병증이 발생하는 매우 드문 질환이다[1,2]. 종종 생명을 위협하는 치명적인 출혈 합병증이 발생하는 것으로 알려져 있어, 관련된 증상이 동반된 환자에서 의심을 하여 조기에 진단을 하는 것이 매우 중요하다.
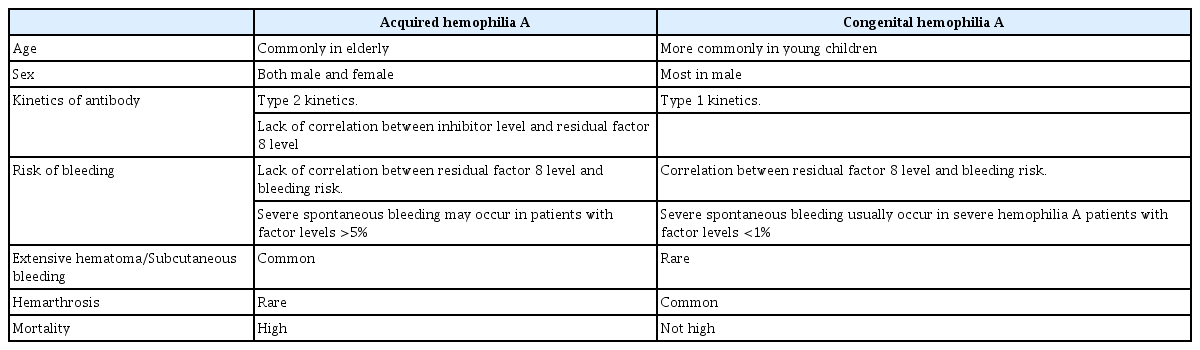
후천성혈우병 A는 100만 명 중 1년에 0.2-1.48명이 발생하는 것으로 보고되고 있다. 20-30대 여성에서는 임신이나 자가면역성 질환과 관련하여 종종 발생하나, 대부분은 65세 이상의 고령(중앙값 64-78세)에서 남녀에 모두 비슷한 비율로 발생한다[1-4]. 소아에서는 매우 드문 것으로 알려져 있다. 자가면역성 질환(전신홍반루푸스, 류마티스관절염 등), 악성종양, 임신(대부분 첫 번째 임신에서 발생하며, 출산 후 1-4개월에 발생함. 자가항체가 태반을 통과하므로 태아에 출혈 위험이 있음) [5,6], 감염[7], 약제 등 후천성혈우병 A 발생에 관련된 원인이 있는 경우가 50% 정도 되며, 나머지 50%에서는 원인불명(idiopathic)으로 밝혀진 관련된 질환 없이 발생하는 것으로 알려져 있다[1,8]. 비록 약 30% 정도에서는 출혈이 심하지 않아 출혈에 대한 지혈치료가 필요 없으나, 최근의 대규모 보고에 따르면 94.6%에서는 출혈성 임상양상이 동반되었고, 77%의 자연출혈과 70%의 심각한 출혈(혈색소 <8 g/dL 또는 혈색소 >2 g/dL 감소로 정의됨)이 관찰되었다[3]. 선천성혈우병과는 나타나는 임상양상에 차이가 있다. 선천성혈우병에서 주로 관찰되는 관절강내출혈(hemarthrosis)은 드물고, 후천성혈우병 A에서는 피하출혈이 흔하게 관찰된다(> 80%). 근육내 출혈이나 위장관계 출혈도 종종 관찰된다(Table 1) [1,2,4]. 사망률은 특히 65세 이상의 고령이나 기저 악성종양을 동반한 경우에는 20% 이상으로 보고되고 있다[2]. 기저질환 관련 사망이 많고, 출혈에 의한 사망은 5-10% 정도로 알려져 있다(이전 연구에서 3.2%와[3] 9.1%로[4] 보고됨). 후천성혈우병 A는 진단의 지연으로 인하여 치명적인 출혈에 의한 사망이 발생할 수 있으며, 진단과 적절한 초기 조치가 되지 않은 상태에서 출혈 조절을 위한 추가 시술 중에 출혈 합병증이 더 악화될 수 있으므로, 적절한 진단과 치료에 대한 교육이 매우 중요한 질환이라 할 수 있다.
Comparison of acquired hemophilia A & congenital hemophilia A
진단
과거에 출혈 경향이 없었고, 출혈 질환에 대한 가족력이 없는 환자에서 갑자기 발생하거나 가벼운 시술이나 상처 후에 심한 출혈이 발생하는 경우에는 후천성혈우병 A를 반드시 의심해 보아야 한다. 진단을 하는 데 초기에 가장 중요한 검사 소견은 프로트롬빈시간(prothrombin time)은 정상이고, 활성화부분트롬보플라스틴시간(activated partial thromboplastin time, aPTT)이 연장이 있으면서, 혼합검사(mixing test, 정상인의 혈장과 같은 양의 환자의 혈장을 넣어서 환자의 연장된 aPTT의 교정 정도를 확인하는 검사)에서 환자의 연장된 aPTT가 50% 이상 교정이 되지 않는 결과이다[9]. 응고인자 8번의 불활성화는 시간과 온도에 의존적이기 때문에 후천성혈우병 A 진단을 위한 혼합검사 판독에 있어 상온에서 즉시 검사 결과를 판단하면 안 되고, 37°C에서 1-2시간 후에 결과를 판독해야 한다[9]. 루푸스항응고물질(lupus anticoagulant, LA)이 검출되는 항인지질항체증후군(antiphospholipid antibody syndrome)에서도 연장된 aPTT 검사 결과와 혼합검사에 연장된 aPTT가 교정이 되지 않는 결과가 나오므로 후천성혈우병 A를 진단하기 위해서는 LA 음성임을 확인해야 한다. 항인지질항체증후군에서의 LA는 혼합검사에서 보통 상온에서 바로 교정이 안 되는 결과가 나오므로 후천성혈우병 A와 혼합검사로 대략적으로 감별이 가능하다[2,10].
후천성혈우병 A의 최종 진단은 감소된 혈중 8번 응고인자 농도와 8번 응고인자에 대한 자가항체의 확인으로 가능하다. 후천성혈우병 A에서 관찰되는 자가항체는 주로 면역글로불린G (immunoglobulin G, IgG)이며, 응고인자 8번 경쇄의 C2 domain이나 A2 domain, A3 domain에 대한 직접적인 항체이다. 혈우병에서 응고인자에 대한 항체의 농도는 주로 베데스타 분석법(Bethesda assay)을 사용한다(Nijmegen 방법이 민감도를 올릴 수 있음). 항체의 양을 정량화할 때 1 Bethesda Unit (BU)은 8번 응고인자 활동도를 50% 비활성화시키는 항체의 양으로 정의된다. 그러나, 후천성혈우병 A의 자가항체는 선천성혈우병 환자에서 발생하는 8번 응고인자에 대한 항체에서 보이는 1형 역동학(kinetics)과는 달리 2형 역동학을 가지고 있어서(non-linear complex inactivation pattern, 비선형 복합체 불활성화 양상), 초기에 8번 응고인자의 활동도를 빠르게 비활성화시키나 이후에는 천천히 작용하기 때문에 베데스타 분석법으로 측정한 8번 응고인자에 대한 자가항체의 농도로 8번 응고인자에 대한 비활성 강도를 정확히 예측하기 어렵다[10]. 즉, 측정된 잔류 8번 응고인자의 농도로 출혈의 정도를 예측하기 어렵다. 그러므로, 선천성혈우병과는 달리 5% 이상의 8번 응고인자를 가지고 있는 후천성혈우병 A에서도 치명적인 출혈이 발생할 수 있다(Table 1).
치료
후천성혈우병 A의 치료는 출혈과 관련된 합병증의 예방 및 치료와 응고인자에 대한 항체 제거가 주요 목표가 되어야 한다. 또한 기저 동반질환이 확인된 경우에는 동반질환에 대한 치료도 같이 병행되어야 한다. 출혈의 위험이 있으므로 꼭 필요한 시술이 아니면 피해야 한다.
출혈과 관련된 합병증의 치료
후천성혈우병 A에서 25-30%에서는 출혈관련 합병증이 심하지 않아서 이에 대한 지혈치료가 필요하지 않다. 측정된 잔류 8번 응고인자 농도나 자가항체의 농도로 출혈의 정도를 예측하기 어려우므로 임상적인 출혈 양상에 따라 적절한 응고치료가 필요하다[1,2,8].
후천성혈우병 A 환자에서는 8번 응고인자에 대한 중화 자가항체가 있으므로 8번 응고인자의 응고기전을 우회하는 우회치료(bypassing therapy)가 필요하다. 우회치료로 활성화된 프로트롬빈 복합체 농축물(Activated prothrombin complex concentrate, aPCC [FVIII inhibitor bypassing activity, FEIBA, Baxalta, Bannockburn, IL, USA]) 50-100 units/kg을 8-12시간마다 천천히 정주하거나(최대 용량 200 U/kg/24 hrs), 재조합 활성화 7번 응고인자(Recombinant activated factor VII, rFVIIa [NovoSeven, Novo Nordisk, Bagsvaerd, Denmark]) 70-90 μg/kg을 2-3시간마다 지혈이 될 때까지 투약한다. 지혈이 어느 정도 된 이후에는 투약 간격을 늘릴 수 있다[1,2]. 유럽에서 진행된 대규모 European Acquired Hae mophilia (EACH2) registry 연구에서 출혈을 치료하기 위하여 사용된 약제는 rFVIIa (56.7%), aPCC (20.5%), 사람재조합 8번응고인자(18.2%), 데스모프레신(desmopressin, DDVAP, 4.6%)으로, rFVIIa나 aPCC를 사용한 우회치료로 91.8%에서 출혈을 조절할 수 있었다. 사람재조합 8번 응고인자를 사용한 경우에는 낮은 출혈 조절률을 보였다(69.6%) [11]. rFVIIa나 aPCC를 사용한 우회치료를 시행할 경우 동맥이나 정맥의 혈전증의 위험이 증가할 수 있음을 고려해야 한다(EACH2 registry 연구에서 보고된 혈전증 빈도; rFVIIa 2.9%, aPCC 4.8%) [11]. 특히 고령에서 암을 동반하고 있거나 이전에 혈전증의 과거력이 있는 경우 주의해야 한다. aPCC 치료에 반응이 적절하지 않은 경우에는 rFVIIa으로 치료약제를 변경하여 시도해 볼 수 있다. 후천성혈우병 A 환자에서 주요 시술이나 수술이 필요한 경우에는 예방적으로 이들 우회치료약제를 사용해야 한다[12]. 이러한 우회치료약제를 사용할 수 없는 상황에서만 재조합 8번 응고인자나 혈장유래 사람 응고인자 8번 농축물을 사용하거나13 근거가 미약하지만 데스모프레신을 사용해 볼 수 있다. 주로 8번 응고인자에 대한 자가항체 농도가 매우 낮은 경우에 주로 반응을 기대할 수 있다. 점막출혈이 있는 경우에는 신장 관련 출혈이 없다면 우회치료약제에 추가로 항섬유소용해제(antifibrinolytic agents; tranexamic acid)를 추가로 사용해 볼 수 있다[2].
우회치료약제에 대한 반응을 평가하는 표준화된 검사법이 없으므로 출혈에 대한 치료에 대한 반응 평가는 임상적으로 결정해야 한다. 8번 응고인자 농도가 회복이나 aPTT가 정상화되는 것을 참고할 수는 있지만 근거가 부족하므로, 임상적인 출혈 양상을 보고 우회치료약제 치료 반응을 평가하고 우회치료약제 치료 중단을 결정해야 한다.
응고인자에 대한 항체 제거
후천성혈우병 A 환자에게 출혈에 대한 합병증 치료에 추가로 발생한 8번 응고인자에 대한 자가항체를 제거하기 위한 면역억제치료가 권고된다. 치료를 하지 않아도 약 30% 정도에서는 자연적으로 항체가 없어진다고 알려져 있다. 특히 자가항체의 농도가 낮은 경우, 출산 후, 약제 관련 후천성혈우병 A 환자에서 저절로 항체가 소실된다고 알려져 있다. 그러나, 이들 환자에서도 치명적인 출혈이 발생할 수 있으므로, 이에 대한 위험을 줄이기 위해서는 모든 성인 후천성혈우병 A 환자에게 면역억제치료가 즉시 권고된다. 면역억제치료로 적절한 약제를 선택하기 위해서는 그동안 발표된 대규모 연구들을 검토해 보아야 한다. 영국에서 시도된 UKHCDO (United Kingdom Haemophilia Centre Doctors’ Organisation) 감시 연구에서는[4] 스테로이드 단독치료(보통 매일 prednisolone 1 mg/kg)와 스테로이드와 세포독성 약제(보통 매일 cyclophosphamide 1-2 mg/kg)의 병용치료에 있어 완전관해 유도까지의 기간에 차이는 없었다(중앙값; 49일 vs. 39일). 유럽에서 진행된 EACH2 registry [14] 연구에서 1차 치료로 스테로이드 단독, 스테로이드+cyclophosphamide, 그리고, rituximab-기반 치료를 비교하였다. 스테로이드+cyclophosphamide 치료군에서 더 높은 완전관해율(80%)과 더 짧은 관해 유도기간(40일)을 보고하였다(스테로이드 단독군; 58%, 32일, rituximab-기반 치료군; 61%, 64일). 그러나, 생존율에서는 이들 3군에 차이를 보이지 않았다. Acquired Hemophilia Working Group of the German, Austrian and Swiss Thrombosis and Hemostasis Society (GTH-AH) registry [15]의 전향적 연구(n=102)에서는 우선 1차 치료제로 스테로이드 단독(60 kg 미만에서는 매일 prednisolone 75 mg/일, 60-100 kg은 100 mg/일, >100 kg에서는 150 mg/일) 치료를 3주간 시도하고, 부분관해에 도달하지 못하는 경우 스테로이드에 경구용 cyclophosphamide (60 kg 미만에서는 100 mg/일, 60-100 kg은 150 mg/일, >100 kg에서는 200 mg/일)를 추가하였고(4-6주째, n=35), 이후에도 반응이 없으면 스테로이드에 rituximab (375 mg/m2/주, 4주간)을 추가하였다(7-10주째, n=12). 부분관해(정의; FVIII>50 IU/dL, 활동성 출혈 없음, 출혈억제를 위한 응고치료약제 사용이 24시간 이상 없음)는 83%에서 도달하였고(부분관해 도달일 중앙값; 31일), 완전관해(정의; 부분관해+자가항체 음성, 감량된 prednisolone 용량 <15 mg/일, 다른 면역억제제 치료 중단)는 61% 환자에서 관찰되었다(완전관해 도달일 중앙값; 79일). 1년 생존율은 68%이었다. 후천성혈우병 A 진단 시의 낮은 혈중 8번응고인자 농도(<1 IU/dL)를 가진 경우 치료에 따른 완전관해율이 낮았다. 베데스타 분석법으로 측정한 항체에 대한 BU값보다는 효소면역측정법(Enzyme-Linked Immuno-Sorbent Assay, ELISA)으로 측정한 8번 응고인자에 대한 면역글로불린G가 높은 경우 치료에 대한 반응이 불량하였다[16]. 또한, 진단 시의 8번 응고인자에 대한 면역글로불린A가 높은 경우와 불량한 전신상태(WHO 전신상태 < 2)를 보인 환자군에서 불량한 치료반응과 짧은 생존율을 보였다[17]. 진단 시 악성종양을 동반한 경우에도 다른 동반질환을 가진 경우보다 짧은 생존율을 보였다[15,17].
결론적으로 후천성혈우병 A 환자의 1차 면역억제치료로는 스테로이드 단독치료가 가능하며, 비록 장기 생존율의 이득은 확인되지 않았지만 스테로이드+경구용 cyclophosphamide 치료도 1차 치료로 시도해 볼 수 있을 것으로 판단된다. 이들 치료에 반응이 적절하지 않은 경우에는 rituximab-기반 치료를 시도해 볼 수 있다. Rituximab 이외에도 mycophenolate mofetil, azathioprine, vincristine 그리고 cyclosporine 등을 이용한 면역억제 치료도 고려해 볼 수 있다. 고용량의 면역글로불린 주사(intravenous immunoglobulin)는 권고되지 않는다. 면역억제치료 중 감염의 위험이 증가하고, 이와 관련된 사망도 보고되고 있어서 면역억제치료 중 보존적 치료에 대한 철저한 감시가 요구된다[1,2,12].
임신과 관련하여 발생한 후천성혈우병 A 환자는 자연적으로 회복되는 경우도 있고 다른 원인에 의한 후천성혈우병 A보다는 전체 생존율이 좋다. 면역억제치료는 산모나 태아에 대한 독성을 고려하여 스테로이드 단독을 1차 치료로 권고하고 있다. Cyclophosphamide는 임신이나 수유 중에 안전하지 않다고 알려져 있다[2,18].
후천성혈우병 A 환자에서 면역억제치료 중에는 치료 반응과 재발 여부 판단을 위해서 매주 8번응고인자 농도와 항체농도를 측정할 것을 권고하고 있고[2], 자가항체가 음전이 되는 완전관해에 도달한 이후에도 15-24%의 재발이 보고되고 있어, 6개월간 매달, 이후 1년까지 2-3개월마다 추적 검사를 권고하고 있다. 완전관해 도달 1년 이후에도 6개월마다 재발에 대한 추적 검사가 필요하다[12].
결론
후천성혈우병 A는 자가면역질환의 하나로 선천성혈우병과 다른 임상양상과 기전을 가지고 있으므로 질환에 대한 정확한 이해와 적절한 진단이 중요하다. 출혈 질환에 대한 과거력이 없는 환자에서 설명할 수 없는 aPTT값의 연장을 보이면서 출혈 경향을 보이는 경우 반드시 후천성혈우병 A를 고려해야 한다. 또한 측정된 잔류 8번 응고인자의 농도로 출혈의 정도를 예측하기 어려우므로 임상적인 출혈 양상에 따라 적극적인 초기 치료가 필요하다. 출혈과 관련된 합병증의 예방 및 치료를 위한 우회치료와 면역억제제를 사용한 응고인자에 대한 항체 제거가 치료의 주요 목표가 되어야 한다. 비교적 드문 질환으로 여전히 표준치료 지침에 대한 자료가 많이 부족한 상황으로 대부분 후향적 연구 결과에 따른 전문가의 권고에 의존하고 있다. 효과적인 치료방법 확립을 위해서는 다기관 연구가 활발히 진행되어야 할 것으로 판단된다.
Acknowledgements
이 논문은 한국혈전지혈학회의 연구비를 지원받아 진행되었음(KSTH 2016-003).